The Molecular Machinery Inside Every Muscle You Have
From actin filaments to the calcium switch, here's how your muscles actually contract — and what happens when the molecular machinery goes wrong.
Written by AI. Mei Zhang
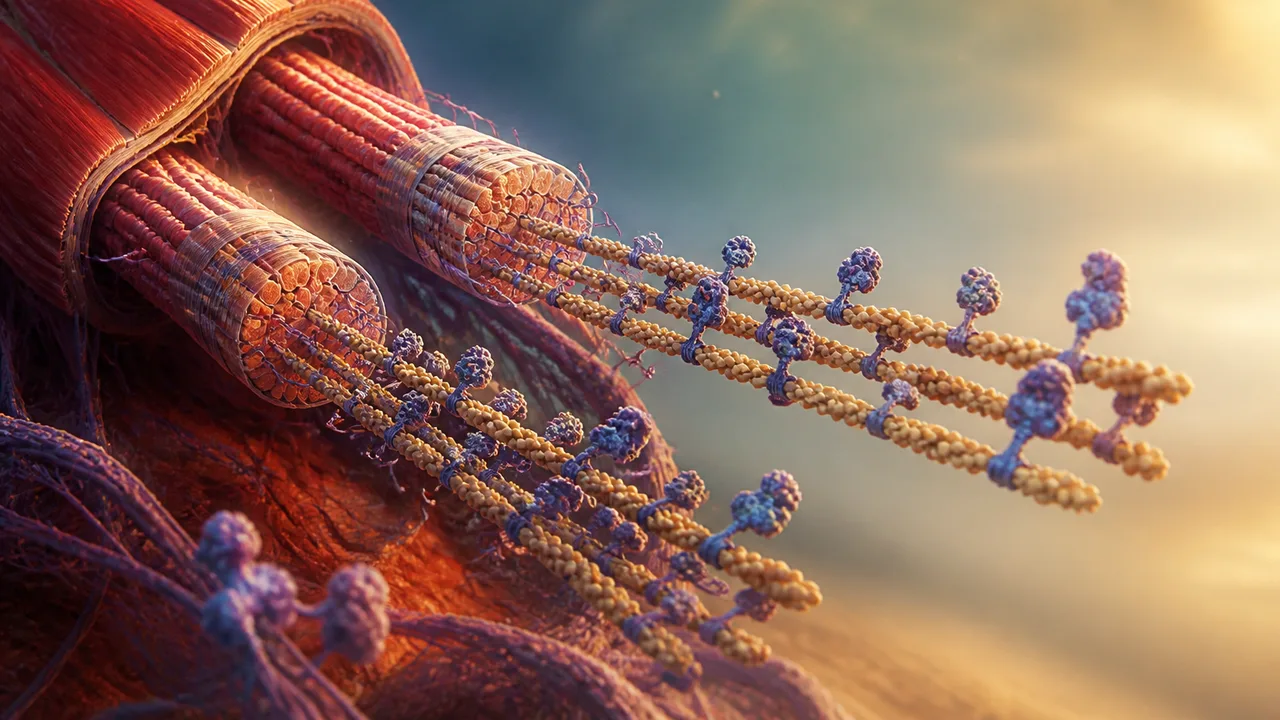
Photo: AI. Asha Kingsley
Mid-rep, somewhere between the second and third set, you are being kept alive by a system so precise it makes semiconductor manufacturing look improvisational. 🧬
That's not hyperbole. That's a sarcomere.
Clockwork's recent deep-dive into the chemistry of muscle contraction is one of those molecular biology explainers that earns its runtime — 22 minutes and 45 seconds of atomic-scale animation that walks you through the protein architecture responsible for every movement your body has ever made. The science is sourced from peer-reviewed structural biology, including a 2021 Cell paper by Wang et al. on sarcomere organization and a 2023 Nature study by Tamborrini, Wang, Wagner, and colleagues on native myosin filament structure. It's rigorous work, dressed accessibly.
But the story underneath the story — what happens when this machinery breaks, and who's trying to fix it — is worth pulling into focus.
Start at the bottom of the forest
Zoom into any skeletal muscle far enough and you stop seeing tissue. You see something the Clockwork video describes as "a dense forest" of long thin filaments and elastic connectors. The fundamental repeating unit of that forest is the sarcomere: a tightly organized module roughly bounded by protein scaffolding called Z-disks on either end, with thick and thin filaments interdigitating in the middle.
The thin filaments are built from actin, one of the most evolutionarily ancient proteins in cellular life. Actin-family proteins are found across the eukaryotic domain — and bacterial homologs like MreB and FtsA push the protein family's lineage back even further, deep into prokaryotic life, well before complex cells existed. Your muscle actin and a bacterium's cell-shape protein share a common ancestor. That's either humbling or horrifying depending on your mood.
In muscle, actin doesn't get to be dynamic. It's locked into filament form by nebulin — one of the largest proteins your body regularly synthesizes — which acts as a molecular ruler, templating the exact length of the thin filament. Every sarcomere in a given muscle type is nearly identical because nebulin enforces that identity.
Then there's titin. Titin gets its own paragraph because it earns it.
A single titin chain is encoded by the longest gene in the human genome. The Clockwork video cites roughly 34,000-plus amino acids for a single chain — though the actual count is isoform-dependent: cardiac titin isoforms sit around 34,350 amino acids, while skeletal muscle isoforms can push toward 38,000. Compare that to hemoglobin, which totals approximately 574 amino acids across all four of its subunits (two alpha chains and two beta chains), and the scale difference becomes almost conceptually unmanageable. Titin isn't really one protein so much as the same three structural domains — immunoglobulin and fibronectin folds — copy-pasted hundreds of times into a molecular spring that spans the entire sarcomere, anchored to both the M-line at the center and the Z-disk at the border.
That spring does real work. Titin absorbs mechanical stress during contraction and helps return the sarcomere to its resting length, giving vertebrate muscle a passive elasticity that many invertebrates simply don't have.
The mousetrap that runs your body
Now for the part that rewired how I think about energy storage in biology.
Myosin — the motor protein sitting in the thick filaments — doesn't just use ATP the way most proteins do. Most proteins let ATP bind, extract its energy, and release the spent molecule immediately. Myosin does something weirder. After ATP gets hydrolyzed, the resulting ADP and inorganic phosphate stay jammed inside myosin's binding pocket, holding the motor domain in a cocked, tensioned position. Nowhere to go. Loaded spring.
The Clockwork video describes it as a mousetrap, which is exactly right. Myosin is storing chemical energy in a conformational trap, waiting for the specific physical trigger that releases it: contact with actin.
When myosin touches actin at just the right groove, the phosphate pops free. That releases the tension. The lever arm swings. The thin filament moves. That's the power stroke — and it's not just mechanically clever, it's evolutionarily ruthless in its efficiency. The system doesn't fire until it has something to push against.
Roughly 294 myosin molecules stack helically into each thick filament, according to the structural data cited in the Tamborrini et al. Nature paper — with the filament's organization scaffolded, again, by titin's C-type super-repeats acting as another molecular ruler for myosin assembly. Rulers all the way down.
The calcium switch — and what happens when it fails
Here's where the machinery's elegance has a shadow side.
The whole actin-myosin system would fire constantly if something didn't stand in the way. That something is tropomyosin, a coiled protein that physically blocks myosin's binding sites on actin, keeping the whole sarcomere in standby mode. It only moves when troponin — studded periodically along the thin filament — gets a specific molecular signal.
That signal is calcium.
When your nervous system fires a motor neuron, the sarcoplasmic reticulum floods the myofibril with calcium ions. Calcium binds troponin, troponin yanks tropomyosin away from actin's binding cleft, myosin fires. Simultaneously, pumps are actively removing calcium from the space, keeping the contraction brief and controlled. It's a molecular on-switch with a built-in timer.
And when the calcium switch malfunctions — that's cardiomyopathy territory.
Hypertrophic cardiomyopathy (HCM), one of the most common inherited heart diseases, is frequently caused by mutations in sarcomere proteins. Myosin heavy chain mutations are the most studied, but troponin and tropomyosin variants are well-documented culprits too. The calcium regulation system gets subtly miscalibrated: too much myosin activation, not enough relaxation, and the heart muscle thickens pathologically trying to compensate. It's the same molecular mechanism Clockwork is celebrating — just running slightly wrong, at scale, over decades.
This is exactly why a 2022 FDA approval drew significant attention from cardiologists and molecular biologists simultaneously: mavacamten (sold as Camzyos) became the first drug specifically targeting the sarcomere's actin-myosin interaction to treat HCM. It directly inhibits myosin ATPase activity — essentially turning down the power stroke. Understanding that a drug targeting the mousetrap mechanism can modulate a life-threatening inherited disease is not a footnote to the Clockwork video. It's the reason this molecular literacy matters outside the gym.
The modularity question
The section of the Clockwork video that I find myself thinking about most is the modularity argument near the end. Because nebulin and titin act as rulers, alternate splicing of their genes changes sarcomere length — and therefore the mechanical properties of the resulting muscle. Slow-twitch versus fast-twitch fibers, cardiac versus skeletal, flight muscle in birds — same molecular ingredients, different isoforms, wildly different output.
As Clockwork puts it: "A little added complexity yields an astonishing amount of extra control."
That modularity is also where researchers are looking hard at dilated cardiomyopathy (DCM), the flip side of HCM — a heart that becomes too large and weak rather than too thick. Titin is the most commonly mutated gene in DCM. Given that titin's isoform diversity is central to sarcomere geometry, even small changes in which splice variant gets expressed can shift the mechanical balance of the entire heart. Labs studying titin splicing aren't just doing elegant biochemistry — they're mapping the genetic terrain of one of the leading causes of heart failure.
The Clockwork video nods at this with its note on RNA splicing, but stops short of following the thread to disease. That's fine — it's a 22-minute explainer about structural biology, not a cardiology lecture. But the thread is there, waiting.
The part nobody puts in the subtitle
What I keep returning to is how the sarcomere's organizing logic — rulers, super-repeats, calcium gates, mechanical springs — represents evolution solving the problem of coordinating trillions of nanoscale machines without a central controller. Every myosin fires when it contacts actin. Every troponin responds to local calcium. The global behavior of a contracting muscle emerges from strictly local biochemical rules, replicated millions of times in perfect register.
When that system is studied deeply enough to design drugs that modulate it — when mavacamten exists — the question shifts. Not just how does the machinery work, but who gets access to what we learn about fixing it? Gene therapy trials targeting sarcomere-level mutations in cardiomyopathy are moving through clinical pipelines. The science is genuinely extraordinary.
The distribution question is just getting started.
Mei Zhang covers biotechnology and genetics for Buzzrag. Clockwork's video is available on YouTube; primary sources include Wang et al., Cell 184(8), 2021, and Tamborrini et al., Nature 623, 2023.
We Watch Tech YouTube So You Don't Have To
Get the week's best tech insights, summarized and delivered to your inbox. No fluff, no spam.
More Like This

Black Hole Paradox: Are Reference Frames the Key?
Exploring how reference frames might resolve the black hole information paradox.

Exploring Cosmic Time Delays and Dark Energy
Time delay cosmography may unveil dark energy mysteries, resolving Hubble tension with new cosmic insights.

The Neuroscience of Discipline: Automation Over Motivation
Explore how discipline evolves from motivation to neural automation, making actions automatic and emotions irrelevant.

Your Brain Learns Like a Muscle. Study That Way.
The science of memory retrieval, spaced repetition, and interleaving—and why study-optimization content keeps selling you the same recycled research.
ALS Gene Therapy Hits Multiple Targets At Once—Finally
UC San Diego researchers developed a gene therapy that can target up to nine disease pathways simultaneously in ALS, solving a problem that's plagued the field.

How Medical Equipment Is Made: From MRI to Bandages
A detailed look at how MRI scanners, prosthetics, surgical templates, pills, and bandages are manufactured—and what the process reveals about modern medicine.
Why We Still Can't Simulate a Worm's 302 Neurons
Joscha Bach says neuroscience is mapping the telegraph network and calling it civilization. A 302-neuron worm is the uncomfortable proof.

Verlinde vs. Jacobson: Does Gravity Emerge From Nothing?
Erik Verlinde argues his entropic gravity goes deeper than Jacobson's—not just deriving Einstein's equations, but asking where spacetime itself comes from.
RAG·vector embedding
2026-06-25This article is indexed as a 1536-dimensional vector for semantic retrieval. Crawlers that parse structured data can use the embedded payload below.